Activities Report / InformationREPORT & INFORMATION
2025.09.01
Medical and disease
information
- #Lysosomal storage diseases
- #Mucopolysaccharidosis
- #MPS
- #Overview
Basic Knowledge of Mucopolysaccharidoses (MPS)
- What are Mucopolysaccharidoses?
-
Mucopolysaccharidoses (MPS) are a group of disorders collectively known as lysosomal storage diseases.
Mucopolysaccharides (glycosaminoglycans or GAGs) are components of the body with a chain-like structure of sugars, as shown in the diagram.
For example, chondroitin, a type of mucopolysaccharide commonly found in cosmetics and supplements, plays an important role in the extracellular matrix – the space between cells – where it functions as both a lubricant and an adhesive.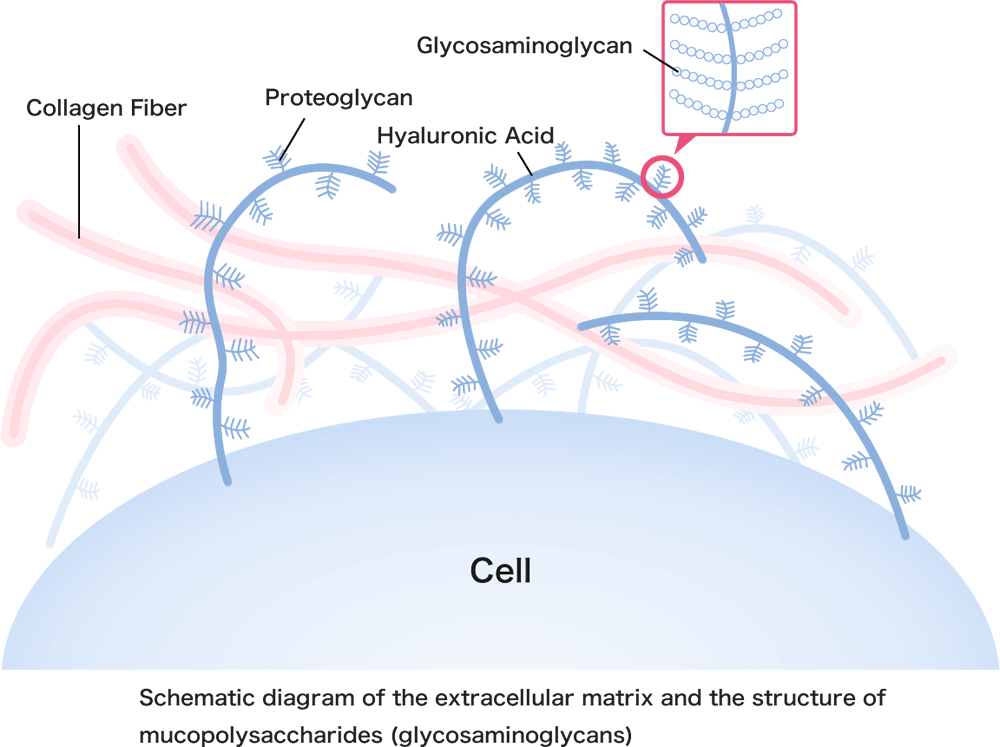
When an individual is born with a deficiency or reduced enzyme activity that breaks down mucopolysaccharides, these substances gradually accumulate in the body, leading to the development of MPS.
- Types of MPS
-
MPS includes several types, which are mainly classified by the specific enzyme deficiency responsible for the condition.
Major Types of MPS
Type Another Name Affected Enzyme Accumulated Substance MPS I Hurler syndrome α-L-iduronidase DS,HS Hurler-Scheie syndrome Scheie syndrome MPS II Hunter syndrome Iduronate-2-sulfatase DS,HS MPS III Sanfilippo
syndromeType A Heparan N-sulfatase HS Type B α-N-acetylglucosaminidase Type C Acetyl-CoA: α-glucosaminide N-acetyltransferase Type D N-acetylglucosamine-6-sulfatase MPS IV Morquio syndrome Type A N-acetylgalactosamine-6-sulfatase KS Type B β-galactosidase MPS VI Maroteaux-Lamy syndrome N-acetylgalactosamine-4-sulfatase DS,CS MPS VII Sly syndrome β-glucuronidase DS,HS,CS DS: dermatan sulfate, HS: heparan sulfate, CS: chondroitin sulfate, KS: keratan sulfate
Note: Besides the main types, rarer forms such as MPS IX and MPS-plus syndrome have also been reported.
- Symptoms
-
The symptoms of MPS can generally be thought of in two groups: those common to all types and those that tend to vary by type.
1)Common Symptoms
Some symptoms, such as distinctive facial features, a large head, or recurrent ear infections (otitis media), can be seen in all types of MPS. However, the severity and timing of these symptoms can vary.
Common symptoms in infancy and early childhood often include: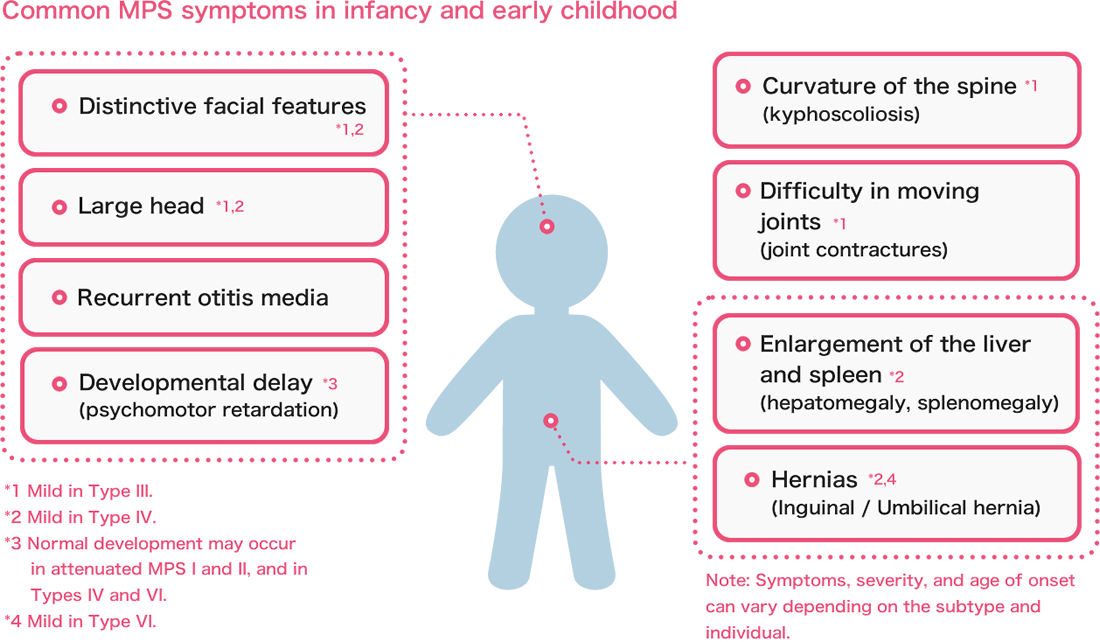
As children grow older, additional symptoms such as short stature, hearing loss, and vision problems may also become apparent.
2)Symptoms Specific to Each Type
Type Key Characteristics of symptoms MPS I - In addition to the common MPS symptoms, corneal clouding, and widespread Mongolian spots may be observed.
- Milder forms typically show little to no developmental delay and have milder physical symptoms.
MPS II - Symptoms are similar to MPS I but typically do not include corneal clouding.
- Non-neurological forms (attenuated MPS II) typically show little to no developmental delay and have milder physical symptoms.
MPS III - Central nervous system symptoms, such as developmental delays and sleep disturbances, are the main features. Physical symptoms are usually less pronounced.
MPS IV - Bone and joint symptoms are the main features. Intellectual development is minimally affected.
MPS VI - Symptoms are similar to MPS I. Intellectual development is minimally affected.
MPS VII - Symptoms are similar to MPS I.
Related Link: Types of mucopolysaccharidoses (NIH)
Go to external site
Please note that the linked websites are not operated by MEDIPAL Group.
To the external sites - Diagnosis
-
When characteristic symptoms of MPS are observed, various diagnostic tests may be performed to confirm the diagnosis. These include X-rays, urine tests to measure glycosaminoglycans (GAGs) levels, enzyme activity assays using blood or cultured skin cells, and genetic testing.
Patients with MPS excrete higher levels of GAGs in their urine. The types of GAGs that accumulate vary depending on the MPS type, and these patterns can help with diagnosis.
For example, dermatan sulfate and heparan sulfate accumulate in MPS I and II, leading to increased proportions of these substances relative to other GAGs in the urine.The likely MPS type is first predicted based on the patient’s symptoms and the pattern of GAGs in their urine. Enzyme activity assays and genetic testing follow this to confirm the diagnosis.
In recent years, expanded newborn screening, which is self-funded and primarily targets MPS types I and II, has started to be offered across various regions in Japan.
- Treatment
-
Based on the patient’s age, MPS type, and disease severity, enzyme replacement therapy (ERT) or hematopoietic stem cell transplantation (HSCT), especially for MPS type I, is considered.
Additionally, symptomatic and supportive care are essential for managing the patient’s symptoms.For a more detailed explanation of treatment approaches, please refer to the following article:
Related Article: Basic Knowledge of Lysosomal Storage Diseases-Treatment

- Prognosis
-
MPS is a progressive and often severe condition for many patients.
However, early intervention has been shown to improve outcomes for treatable types of MPS.Editorial supervision: Kimitoshi Nakamura, MD, PhD (Professor, Department of Pediatrics,
Graduate School of Medical Sciences, Kumamoto University)


Reference materials
- Yoshikatsu Eto edition. Lysosomal Storage Disease - Recent Advances in Pathophysiology, Diagnosis, and Treatment - Revised 2nd Edition. SHINDAN TO CHIRYO SHA, Inc. 2023.
- Information Center for Specific Pediatric Chronic Diseases, Japan. (https://www.shouman.jp/) (Accessed Aug 1, 2025)
- Mucopolysaccharidoses. NIH. (https://www.ninds.nih.gov/health-information/disorders/mucopolysaccharidoses) (Accessed Aug 1, 2025)
