Activities Report / InformationREPORT & INFORMATION
2025.02.03
Medical and disease
information
- #Lysosomal storage diseases
- #Overview
Basic Knowledge of Lysosomal Storage Diseases-Overview
1. What Are Lysosomal Storage Diseases?
Lysosomal storage diseases are a group of disorders caused by the dysfunction of lysosomes, leading to the accumulation of unwanted substances in the body.
Lysosomes are small, sac-like structures within cells that break down unwanted substances.
The interior of a lysosome has an acidic pH (4.5 to 5.5) and contains more than 60 distinct enzymes. These enzymes act like scissors, each specialized in breaking down specific substances. The lysosome uses these enzymes to decompose various unwanted materials, such as sugars, proteins, fats, and DNA.
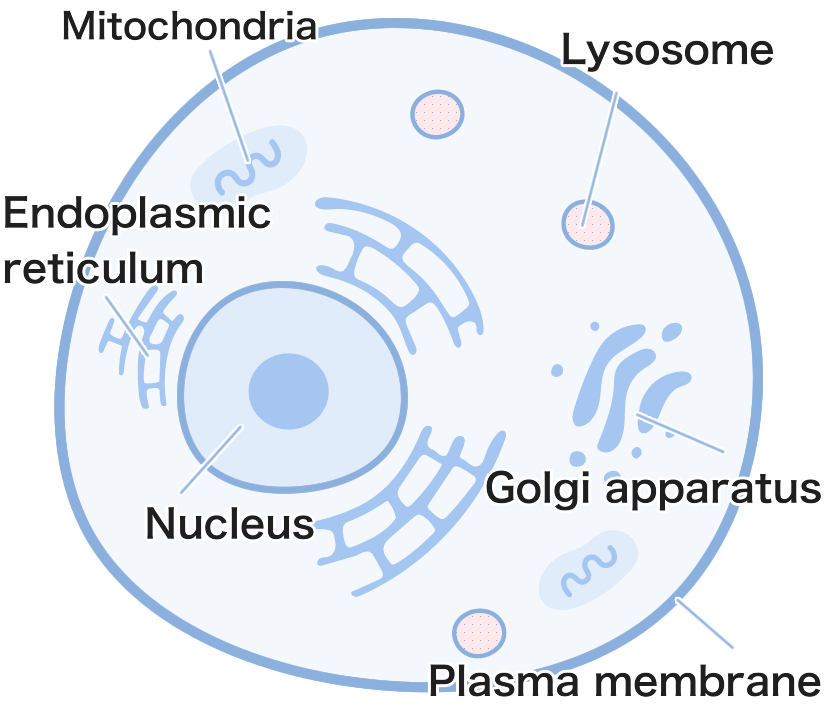 Cell Structure
Cell Structure
The smaller substances resulting from this breakdown are recycled within the body and used to create new materials.
Thus, lysosomes are called the cell’s “waste disposal system” or “recycling center,” playing an important role in maintaining cellular health.

When lysosomes malfunction from birth, substances that should be broken down accumulate, causing what is known as “lysosomal storage diseases.”
2. Causes
Most lysosomal storage diseases result from the dysfunction of enzymes within lysosomes. If a person is born with a reduced activity of a specific enzyme or lacking that enzyme, the substances that the enzyme typically break down will gradually accumulate.
This accumulation is thought to cause various cellular disorders, ultimately leading to lysosomal storage diseases.

3. Typical Conditions
Approximately 60 lysosomal storage diseases have been identified, with the following 31 diseases registered as designated intractable diseases in Japan.
Lysosomal storage diseases registered as designated intractable diseases in Japan (as of December 2024)
- Gaucher disease
- Niemann-Pick disease type A, B / Acid sphingomyelinase deficiency
- Niemann-Pick disease type C
- GM1 gangliosidosis
- GM2 gangliosidosis (Tay-Sachs disease, Sandhoff disease, type AB)
- Krabbe disease
- Metachromatic leukodystrophy
- Multiple sulfatase deficiency
- Farber disease
- Mucopolysaccharidosis type I (Hurler/Scheie syndrome)
- Mucopolysaccharidosis type II (Hunter syndrome)
- Mucopolysaccharidosis type III (Sanfilippo syndrome)
- Mucopolysaccharidosis type IV (Morquio syndrome)
- Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome)
- Mucopolysaccharidosis type VII (Sly syndrome)
- Mucopolysaccharidosis type IX (Hyaluronidase deficiency)
- Sialidosis
- Galactosialidosis
- Mucolipidosis type II, III
- α-Mannosidosis
- β-Mannosidosis
- Fucosidosis
- Aspartylglucosaminuria
- Schindler disease / Kanzaki disease
- Pompe disease
- Acid lipase deficiency
- Danon disease
- Free sialic acid storage disease
- Neuronal Ceroid Lipofuscinosis
- Fabry disease
- Cystinosis
Below is an overview of some typical Lysosomal storage diseases.
Symptoms and severity can vary greatly among individuals, and not all symptoms may be present in a single patient.
Fabry Disease
- Causing Enzyme:
- α-Galactosidase
- Accumulated Substance:
- Globotriaosylceramide
- Disease Types:
- Classic / Late-onset / Heterozygous females *Categorized by age of onset and symptoms.
- Main Symptoms:
-
- Pain, tingling in hands and feet
- Red spots on the skin
- Heart dysfunction
- Stroke
- Sweating less
- Cloudiness in the front part of the eye
- Kidney impairment
- etc.
For more information:
Information Center for Specific Pediatric Chronic Diseases: Fabry Disease
Go to external site
Please note that the linked websites are not operated by MEDIPAL Group.
To the external sitesGaucher Disease
- Causing Enzyme:
- Glucocerebrosidase (also known as β-Glucosidase)
- Accumulated Substance:
- Glucocerebroside
- Disease Types:
- Type I (non-neuronopathic) / Type II (acute neuronopathic) / Type III (chronic neuronopathic) *Categorized by presence and severity of neurological symptoms
- Main Symptoms:
-
- Enlargement of the liver and spleen
- Difficulty stopping bleeding
- Delayed growth
- Anemia
- Bone pain or fractures
- Neurological symptoms etc.
- etc.
For more information:
Information Center for Specific Pediatric Chronic Diseases: Gaucher Disease
Go to external site
Please note that the linked websites are not operated by MEDIPAL Group.
To the external sitesPompe Disease
- Causing Enzyme:
- Acid α-Glucosidase
- Accumulated Substance:
- Glycogen
- Disease Types:
- Infantile-onset / Late-onset (juvenile and adult-onset) *Categorized by age of onset
- Main Symptoms:
-
- Muscle weakness
- Breathing problems
- Heart dysfunction
- Diminished muscle tone
- Delayed growth
- Fatigue etc.
- etc.
For more information:
Information Center for Specific Pediatric Chronic Diseases: Pompe Disease
Go to external site
Please note that the linked websites are not operated by MEDIPAL Group.
To the external sites4. Origin of the Name
Lysosomal storage diseases are named after their discoverers, the deficient enzyme, or the accumulated substances.
The following are examples of typical lysosomal storage diseases and their name origins:
| Disease Name | Origin of Name | |
|---|---|---|
|
Gaucher Disease |
Discoverer |
Philippe Gaucher |
|
Fabry Disease |
William Anderson and Johannes Fabry |
|
|
Pompe Disease |
Johannes Cassianus Pompe |
|
|
Mucopolysaccharidoses |
Accumulated substances |
Mucopolysaccharides |
|
Fucosidosis |
Substances containing fucose |
|
|
Neuronal Ceroid Lipofuscinosis |
Lipofuscin |
|
|
Acid Sphingomyelinase Deficiency |
Deficient enzyme |
Acid Sphingomyelinase |
|
Lysosomal Acid Lipase Deficiency |
Lysosomal Acidic Lipase |
|
5. Classification
Since lysosomal storage diseases include many disorders, various classification methods are used to categorize similar diseases within this group.
1)Classification by Accumulated Substances
The most common method classifies diseases by the type of accumulated substances.
For example, diseases where glycolipids accumulate are classified as “sphingolipidoses,” and those where mucopolysaccharides accumulate are classified as “mucopolysaccharidoses.”

2)Classification by Cause
As our understanding of the molecular biology of lysosomal storage diseases has deepened, a classification method based on the cause of the disease has recently been proposed.
This method classifies diseases based on factors such as enzyme defects, enzyme activator defects, enzyme protection protein defects, enzyme transport defects (enzymes not reaching the lysosome), and substrate transport defects (inability to expel broken-down substances from lysosome).
This new approach contributes to a better understanding of the mechanisms behind lysosomal storage diseases.

Editorial supervision: Kimitoshi Nakamura, MD, PhD (Professor, Department of Pediatrics, Graduate School of Medical Sciences, Kumamoto University)
Reference materials
- Yoshikatsu Eto edition. Lysosomal Storage Disease - Recent Advances in Pathophysiology, Diagnosis, and Treatment - Revised 2nd Edition. SHINDAN TO CHIRYO SHA, Inc. 2023.
- Lysosomal Storage Disease. Japan Intractable Diseases Information Center. (https://www.nanbyou.or.jp/entry/4061) (Accessed Dec 5, 2024)
- Information Center for Specific Pediatric Chronic Diseases, Japan. (https://www.shouman.jp/) (Accessed Dec 5, 2024)
