Activities Report / InformationREPORT & INFORMATION
2024.04.01
Medical and disease
information
- #Lysosomal storage diseases
- #Mucopolysaccharidosis type III
- #Sanfilippo syndrome
Disease Information ‘Mucopolysaccharidosis type III (Sanfilippo syndrome)’
- What is Mucopolysaccharidosis?
-
Mucopolysaccharidosis is one of the diseases classified as “Lysosomal Storage Diseases*”.
Mucopolysaccharidosis occurs when the enzymes needed to break down the Mucopolysaccharides are missing or defective in the cell. The accumulation of mucopolysaccharides causes various symptoms.*Lysosomes are one of the intracellular organelles that work as one of the “waste processing factories” by using various enzymes to break down unnecessary substances in the body. Recently, lysosomes are considered to be involved a variety of cellular functions, not just waste disposal. With Lysosomal storage diseases, enzymes in the lysosomes do not function properly, causing an accumulation of unnecessary and a variety of symptoms. Currently, some 60 diseases have been identified, and 31 of them are registered as designated intractable diseases in Japan.
There are more than 10 enzymes required for the breakdown of mucopolysaccharides. Mucopolysaccharidosis can be classified into several types depending on which enzyme is missing or defective.
For more information, visit “ムコ多糖症.com” by the below link (Japanese only).
ムコ多糖症.com https://mps-jcr.com/ - What is Mucopolysaccharidosis type III?
-
Mucopolysaccharidosis type III (MPS III) occurs when the enzymes needed to break down the heparan sulfate sugar chain are missing or defective.
- Subtypes of MPS III
-
Mucopolysaccharidosis type III is classified into four types A, B, C, and D, depending on which enzyme is deficient. Each enzyme is related to heparan sulfate degradation. The accumulation of heparan sulfate causes various symptoms.
- Subtype
- Affected Enzyme
- Type A
- Heparan N-sulfatase
- Type B
- Alpha-N-acetylglucosaminidase
- Type C
- Acetyl-CoA:alpha-glucosaminide N-acetyltransferase
- Type D
- N-acetylglucosamine 6-sulfatase
- Symptoms
-
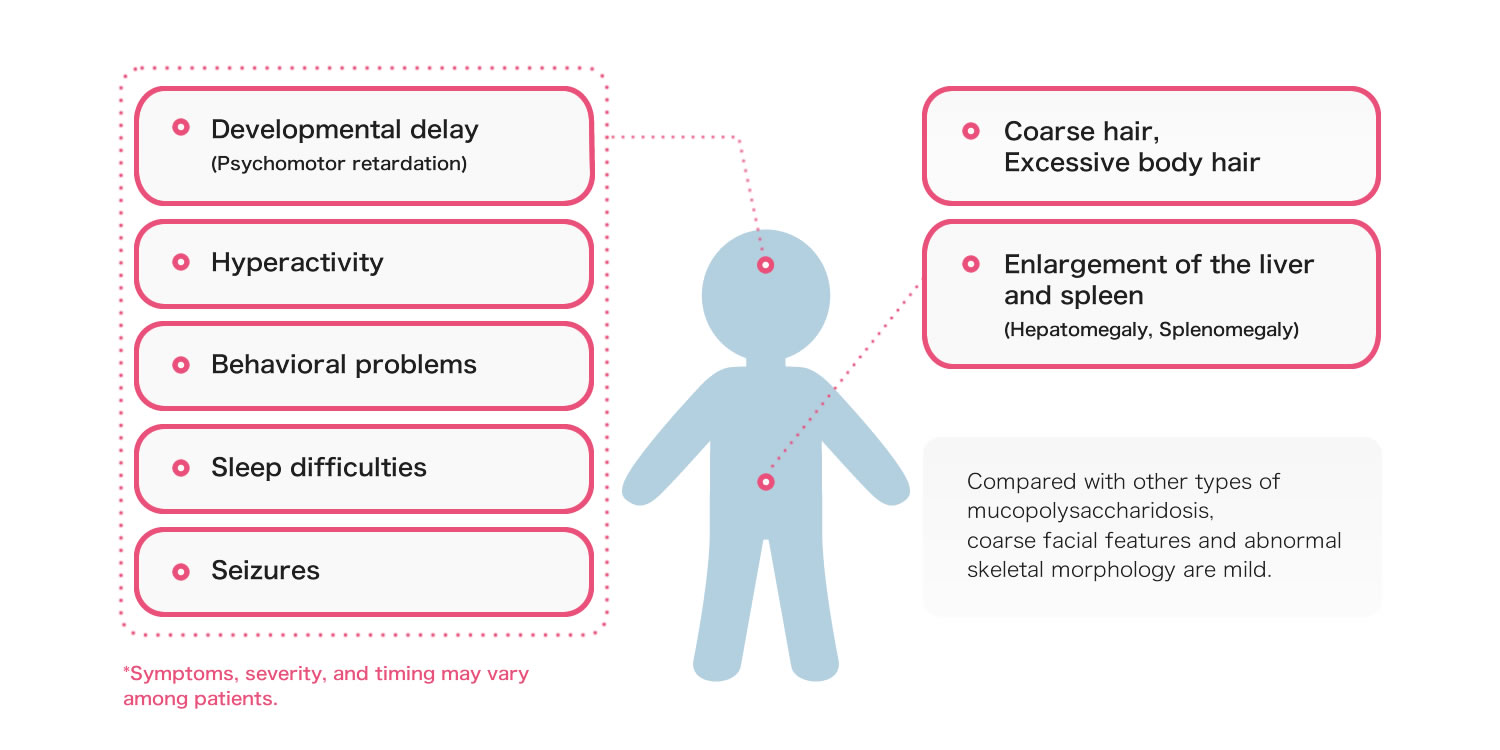
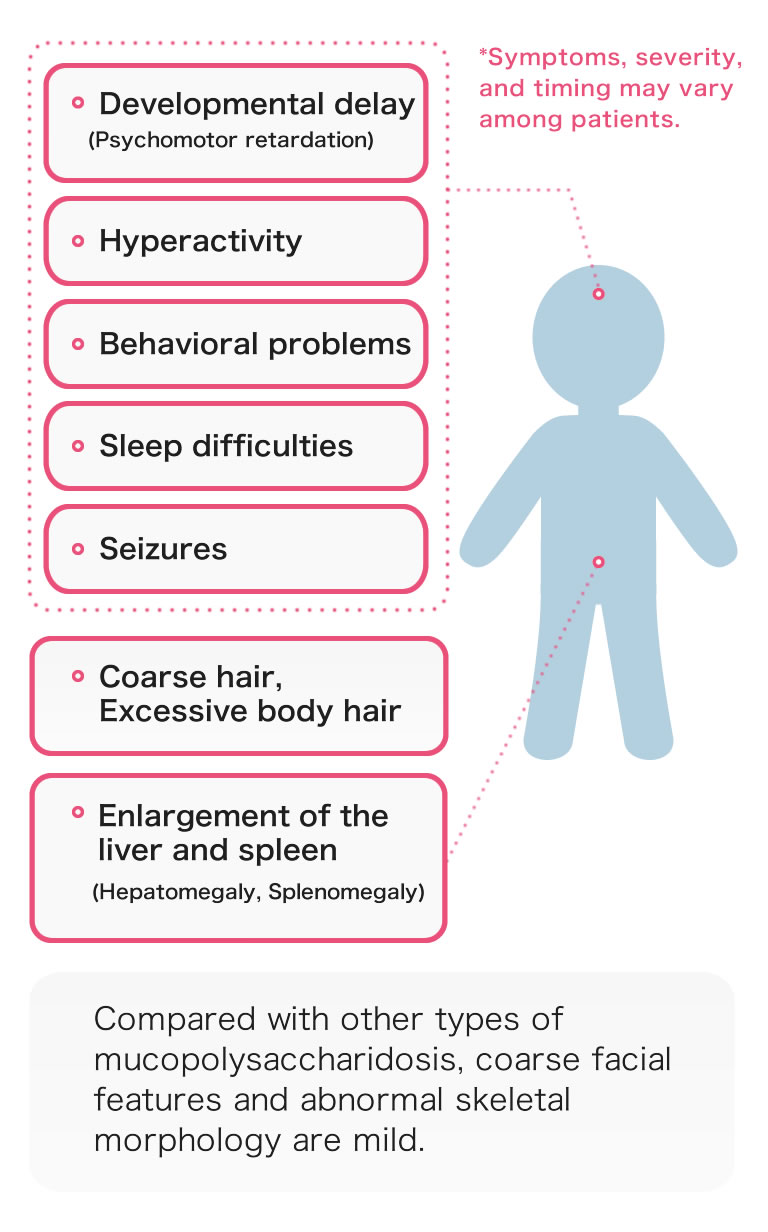
The age of onset and severity vary among patients, but the following symptoms are generally seen in MPS III.
- Diagnosis
-
If there are concerns about mucopolysaccharidosis, the following tests will be performed for determination of mucopolysaccharidosis, X-rays, urinary GAG, leukocyte or fibroblast enzyme activity, and genetic testing.
Since coarse facial features and abnormal skeletal morphology are mild, the definitive diagnosis of MPS III is difficult and often delayed.
MPS type III is often first diagnosed with Autism Spectrum Disorder (ASD) or Attention-Deficit/Hyperactivity Disorder (ADHD) because of delayed speech development and hyperactivity.

- Treatment
-
As of April 2024, there is no approved drug for mucopolysaccharidosis type III. Treatment is tailored to the symptoms, such as the use of anti-epileptic medications for seizures.
- Prognosis
-
In general, lysosomal storage disease progresses gradually as unwanted material accumulates in the cells. MPS III is a progressive and fatal disease that most patients become bedridden in their teens.
For more information about Mucopolysaccharidosis type III, visit “Information Center for Specific Pediatric Chronic Diseases” by the below link.
Information Center for Specific Pediatric Chronic Diseases, Japan Mucopolysaccharidosis Type III https://www.shouman.jp/disease/details/08_06_077/Go to external site
Please note that the linked websites are not operated by MEDIPAL Group.
To the external sites
Go to external site
Please note that the linked websites are not operated by MEDIPAL Group.
To the external sitesEditorial supervision: Yasutsugu Chinen, MD, PhD, Department of Pediatrics, Faculty of Medicine, University of the Ryukyus
Reference materials
- MHLW Intractable Disease Policy Research Project Lysosomal Storage Diseases (including Fabry disease) Research Group Edition. Mucopolysaccharidosis Clinical Manual in accordance with the Diagnostic Guide. SHINDAN TO CHIRYO SHA, Inc. 2015.
- MHLW Intractable Disease Policy Research Project Lysosomal Storage Diseases (including Fabry disease) Research Group Edition. Guide to the Diagnosis of Lysosomal and Peroxisomal Diseases. SHINDAN TO CHIRYO SHA, Inc. 2015.
- Mucopolysaccharidosis type III Information Center for Specific Pediatric Chronic Diseases, Japan (https://www.shouman.jp/disease/details/08_06_077/)(accessed March 1st, 2024)
- Muschol N et al. Sanfilippo syndrome: consensus guidelines for clinical care. Orphanet J Rare Dis. 2022 Oct 27;17(1):391. [PMID: 36303195]
- Delgadillo V et al. Natural history of Sanfilippo syndrome in Spain. Orphanet J Rare Dis. 2013 Dec 6;8:189. [PMID: 24314109]
- Wijburg F A et al. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr. 2013 May; 102(5): 462-470. [PMID: 23336697]
- Rouse C J et al. Mucopolysaccharidosis type IIIB: a current review and exploration of the AAV therapy landscape. Neural Regen Res. 2024 Feb; 19(2): 355-359. [PMID: 37488890]
- Lavery C et al. Mortality in patients with Sanfilippo syndrome. Orphanet J Rare Dis. 2017 Oct 23;12(1):168. [PMID: 29061114]
- Valstar M J et al. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J Inherit Metab Dis. 2010 Dec;33(6):759-67. [PMID: 20852935]
