Activities Report / InformationREPORT & INFORMATION
2024.01.22
Medical and disease
information
- #Lysosomal storage diseases
- #Fucosidosis
Fucosidosis
- What is Fucosidosis?
-
Fucosidosis is one of the diseases classified as “lysosomal storage diseases*.”
Among the lysosomal storage diseases, fucosidosis is known to have a very small number of patients.
There are about a hundred or so cases worldwide, and only a few cases have been reported in Japan.*Lysosomes are one of the intracellular organelles that work as one of the “waste processing factories” by using various enzymes to break down unnecessary substances in the body. Recently, lysosomes are considered to be involved a variety of cellular functions, not just waste disposal. With Lysosomal storage diseases, enzymes in the lysosomes do not function properly, causing an accumulation of unnecessary and a variety of symptoms. Currently, some 60 diseases have been identified, and 31 of them are registered as designated intractable diseases in Japan.
Patients with this disease have the gene mutation in the FUCA1 gene with the ability of α-L-fucosidase to perform its function correctly. The gene mutation in the FUCA1 causes an accumulation of complex sugar molecules in cells that eventually leads cells to malfunction.
- Types of Fucosidosis
-
Fucosidosis is categorized as “Type I” or “Type II” according to age of onset and symptoms.
“Type I” is a severe form with rapid symptom progression characterized by elevated levels of sodium chloride (NaCl) in the sweat, occurring with psychomotor retardation during infancy from 3 months to 1.5 years of age, leading to immobilization.
“Type II” onsets at the age of 1 to 2 years and is characterized by angiokeratomas. It progresses relatively slowly, often into adulthood.
- Symptoms
-
Symptoms and severity vary among patients, but the following symptoms are generally seen.
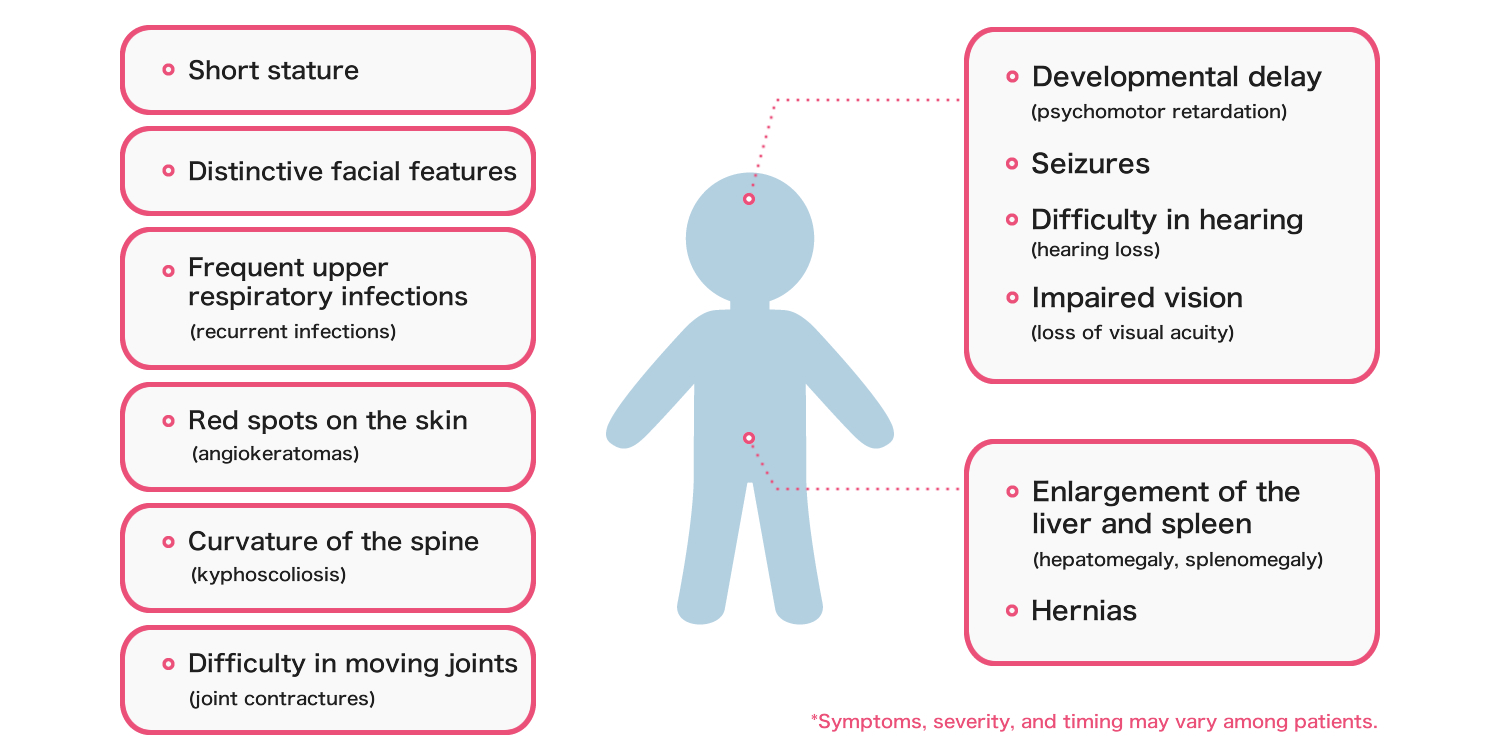
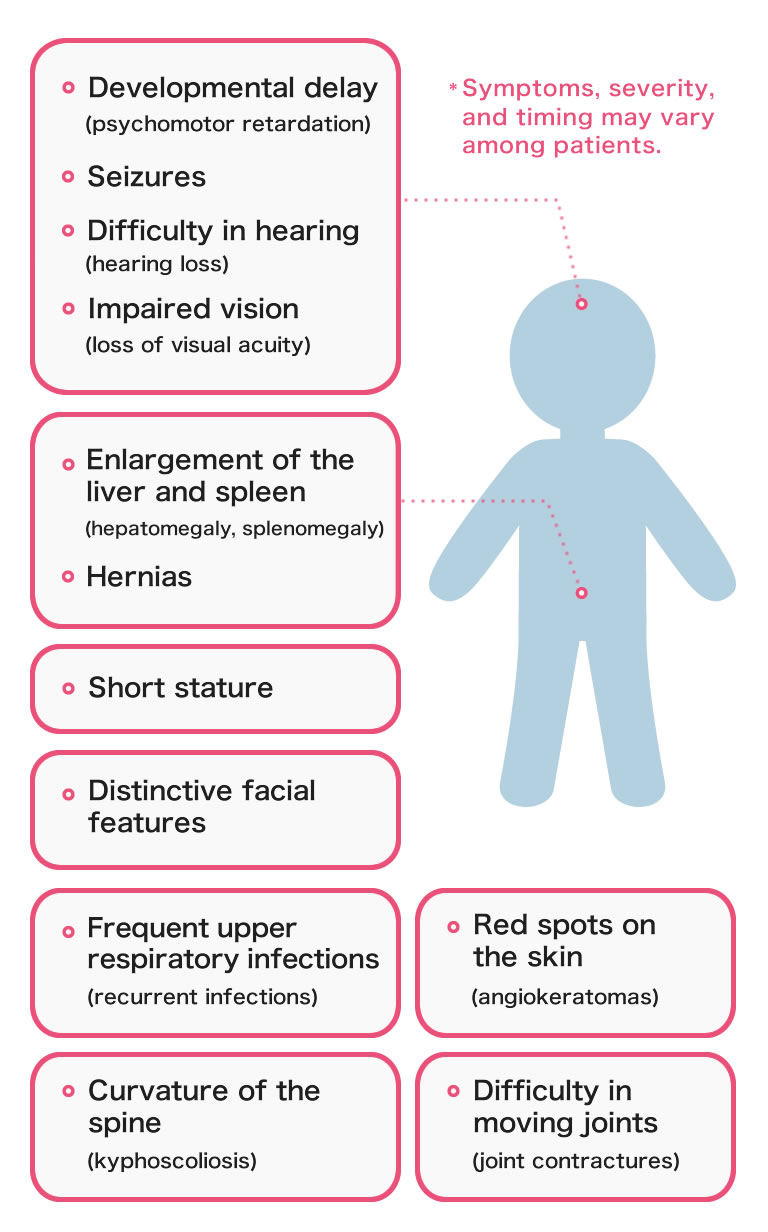
- Diagnosis
-
If there is a possibility of fucosidosis, MRI and urinalysis will be performed.
Enzyme diagnosis and genetic testing using blood and skin cells are then performed to confirm the diagnosis. - Treatment
-
Supportive care is provided based on the symptoms, such as anti-epileptic medications for seizures.
- Prognosis
-
The course of fucosidosis varies by the type of disease, with differences in symptoms even within families.
As symptoms progress, tube feeding or a ventilator may become necessary.
Editorial supervision: Norio Sakai, MD, PhD (Specially Appointed Professor, Developmental Child Health Science, Department of Children's and Women's Health, Division of Health Sciences, Osaka University Graduate School of Medicine)
Reference materials
- MHLW Intractable Disease Policy Research Project Lysosomal Storage Diseases (including Fabry disease) Research Group Edition. Guide to the Diagnosis of Lysosomal and Peroxisomal Diseases. SHINDAN TO CHIRYO SHA, Inc. 2015.
- Fucosidosis. Information Center for Specific Pediatric Chronic Diseases, Japan (https://www.shouman.jp/disease/details/08_06_081/) (accessed August 25, 2023)
- Motohiro Akagi. Fucosidosis. New Area-Specific Syndromes Series No.20, Inborn Errors of Metabolism Syndrome (2nd edition). Nipponrinshosha Co., Ltd. 2012. p.564-567.
- Akagi M et al. Mutation analysis of a Japanese patient with fucosidosis. J Hum Genet. 1999;44(5):323-6. [PMID: 10496076]
- Yoshikatsu Eto edition. Lysosomal Storage Disease - Recent Advances in Pathophysiology, Diagnosis, and Treatment - Revised 2nd Edition. SHINDAN TO CHIRYO SHA, Inc. 2023.
- Willems PJ et al. Fucosidosis revisited: a review of 77 patients. Am J Med Genet 1991;38:111–31. [PMID: 2012122]
- Kaur A et al. Diagnosis and Supportive Management of Fucosidosis: A Case Report. Cureus 2019;11:e6139. [PMID: 31886074]
- Stepien KM et al. Fucosidosis-Clinical Manifestation, Long-Term Outcomes, and Genetic Profile-Review and Case Series. Genes (Basel) 2020;11:1383. [PMID: 33266441]
- Şanlı ME, Uysal S. Fucosidosis: clinical and molecular findings of Turkish patients. Turk J Pediatr. 2022;64(4):795-803. [PMID: 36082656]
